
SMA, czyli rdzeniowy zanik mięśni, to ciężka choroba nerwowo-mięśniowa najczęstsza genetyczna przyczyna śmierci niemowląt oraz małych dzieci. Powodem SMA jest mutacja genu, który produkuje białko niezbędne do utrzymania przy życiu neuronów ruchowych. Kiedy dochodzi do uszkodzenia genu SMN, komórki nerwowe odpowiedzialne za ruch obumierają, a to prowadzi do zaniku mięśni. Do niedawna jedyną pomocą dla chorych na Rdzeniowy Zanik Mięśni była nieco usprawniająca ruchowo rehabilitacja. Pojawił się jednak pierwszy lek, który zatrzymuje postęp choroby. Obecnie nusinersen jest podawany w ramach programu rozszerzonego dostępu w kilkunastu krajach świata. Także w Polsce.
Rdzeniowy zanik mięśni – SMA – to choroba nerwowo-mięśniowa o podłożu genetycznym. 123RF
Co to jest SMA?
Rdzeniowy zanik mięśni jest schorzeniem układu nerwowego i mięśniowego, które objawia się osłabieniem mięśni i upośledzeniem ruchu. Jest to rzadka choroba o podłożu genetycznym, występująca w czterech różnych postaciach, z których każdy pojawia się na różnym etapie życia. Leczenie rdzeniowego zaniku mięśni opiera się przede wszystkim na stosowaniu środków, zwalczających objawy tej choroby.
W Polsce, SMA dotyczy od 400 do 800 chorych – zmagają się z niepełnosprawnością ruchową, oddechową i problemami z mówieniem. Wielu z nich nigdy nie wstało z łóżka. Choroba w żaden sposób nie wpływa na rozwój intelektualny i emocjonalny – chorzy na SMA to osoby inteligentne i otwarte na otoczenie. Niestety, w SMA obumierają neurony w rdzeniu kręgowym odpowiadające za pracę mięśni, na skutek te słabną i stopniowo ulegają zanikowi. Dotyczy to wszystkich grup mięśni – od rąk i nóg, poprzez mięśnie przewodu pokarmowego, aż po te odpowiadające za mowę i oddychanie. Choroba najszybciej postępuje u małych dzieci, w efekcie umierają one z powodu niewydolności oddechowej, spowodowanej przez osłabione mięśnie.
Istnieje 4–5 typów SMA, w zależności od okresu w jakim choroba się ujawnia.
– SMA typu 0 dotyczy noworodków, które w momencie przyjścia na świat nie potrafią oddychać i nie są zdolne do jakiegokolwiek ruchu.
– Objawy SMA typu I pojawiają się w pierwszych sześciu miesiącach życia dziecka. Podstawowymi oznakami są: wiotkość mięśni, zmniejszenie możliwości ruchowych, czasem drżenie języka i palców. Takie dziecko nie dźwiga główki, słabo rusza rękami i nogami, ma kłopoty z oddychaniem. Nie potrafi samo usiąść. Jest to najcięższa postać SMA, najszybciej prowadząca do śmierci.
– W SMA typu II, którego objawy ujawniają się w drugim półroczu życia, dziecko potrafi usiąść, ale nie uczy się chodzić. Zwykle siedzi niepewnie, krzywi kręgosłup, słabiej rusza rękami i nogami.
Jest to najczęściej występująca postać SMA.
– SMA typu III dotyczy dzieci, które już chodziły, a potem zaczynają mieć problem z poruszaniem. Jest to postać najbardziej zróżnicowana ze względu na wiek jak i objawy – te mogą wystąpić, z różną intensywnością, zarówno u 3-, jak i 10-letniego dziecka.
– Na SMA typu IV cierpią dorośli, ale ta postać choroby jest niezwykle rzadka, dotyczy pojedynczych przypadków.
Do niedawna pacjenci byli pozbawieni skutecznej terapii na SMA – mogli korzystać jedynie z rehabilitacji, która w niewielkim stopniu usprawnia ruchowo. W 2016 r. pojawiły się pierwsze badania potwierdzające skuteczność nusinersenu, pierwszego leku, który zatrzymuje postęp choroby, a w wielu przypadkach cofa też jej objawy. Nusinersen został po raz pierwszy zarejestrowany w grudniu 2016 r. przez amerykańską Agencję Żywności i Leków (FDA), a 30 maja 2017 r. przez Europejską Agencję Leków (EMA) i dopuszczony do obrotu przez Komisję Europejską.
Obecnie lek jest podawany w ramach programu rozszerzonego dostępu (EAP) w kilkunastu krajach świata, w tym także w Polsce. Od lutego 2017 r. otrzymuje go 10-osobowa grupa dzieci z Centrum Zdrowia Dziecka w Warszawie, znajdująca się pod opieką prof. Katarzyny Kotulskiej-Jóźwiak. Po 10 miesiącach terapii zostanie przeprowadzona ocena efektywności leczenia. Programem objęta zostanie także grupa kolejnych dzieci z Centrum Zdrowia Dziecka w Warszawie i Uniwersyteckiego Centrum Klinicznego w Gdańsku, a sześcioro dzieci – zakwalifikowanych z województw: śląskiego, małopolskiego, dolnośląskiego i wielkopolskiego – rozpoczęło właśnie terapię nusinersenem w Górnośląskim Centrum Zdrowia Dziecka w Katowicach. Jednocześnie trwają prace nad dalszym rozszerzaniem programu EAP w Polsce.
W ramach programu nusinersen otrzymuje między innymi 11,5-letni Szymon chory na SMA typu I. Jest już po 4 dawkach leku. – Choroba u syna została zatrzymana, z dnia na dzień staje się silniejszy, wyraźniej mówi, lepiej oddycha, zaczął ruszać palcami, lepiej rusza nogami, próbuje kasłać. Powracają ruchy, które miał jako małe dziecko – mówi mama Szymona. Chłopiec rozwijał się prawidłowo do 6. tygodnia, kiedy to pediatra uznał, że dziecko jest nienaturalnie „za wiotkie”. Następnie na podstawie innych objawów neurolog zdiagnozował SMA i skierował Szymona na badania genetyczne – te potwierdziły mutację genu SMN1 odpowiedzialnego za chorobę. Niestety, jedyną dostępną wówczas formą pomocy była fizjoterapia. W ciągu tak długiego czasu, pomimo intensywnej rehabilitacji zaniku mięśni, choroba uległa postępowi, dziecko przestało przełykać i mieć odruch kaszlu. Szymon jest dzieckiem leżącym, nie je samodzielnie, jego oddech wspomaga respirator. Mimo to jest pogodny, ma swoje pasje, a dzięki pomocy taty, który wozi go na zajęcia, uczęszcza do szkoły. Chłopiec posługuje się komputerem za pomocą wzroku. Leczenie Szymona skupia się na uruchomieniu rąk, a cichym marzeniem jego mamy jest, aby w przyszłości mógł usiąść i prowadzić samodzielne życie na wózku.
8-letni Sebastian cierpi na SMA typu II, nieco łagodniejszą postać choroby niż ta, z którą zmaga się Szymon. Chłopiec jeździ na wózku, ale przy pomocy pionizatora potrafi samodzielnie wstać, a dzięki ortezom i specjalnym butom może utrzymać pozycję stojącą. Samodzielnie chodzi do szkoły, a nawet wyjeżdża na obozy wakacyjne. Czasami w nocy musi być dotleniany za pomocą aparatu, a kiedy się przeziębi używa koflatora, który umożliwia kaszel. Niestety, mięśnie rąk ma na tyle osłabione, że potrafi podnieść butelki wody. Choroba spowodowała również, że nigdy nie zaczął chodzić. Z czasem też przestał stać. – Naszym marzeniem jest, aby syn pozostał na wózku i mógł samodzielnie funkcjonować – mówi mama Sebastiana. – A może kiedyś lek przywróci mu możliwość stania… – dodaje.
– Nusinersen jest lekiem zwiększającym efektywność produkcji białka SMN przez gen SMN2, który normalnie produkuje to białko, ale w zbyt małych ilościach. Nusinersen nie leczy SMA, to nie jest naprawa uszkodzonego genu. Powodując jednak wzrost produkcji białka SMN, umożliwia zatrzymanie się choroby, a w niektórych przypadkach poprawę, polepszając komfort życia chorych i ograniczając ich niepełnosprawność. Jest to pierwszy i obecnie jedyny dostępny lek dla chorych na SMA – mówi prof. Katarzyna Kotulska.
Obecnie trwają badania nad innymi lekami na SMA, w tym nad terapiami genetycznymi. – To pokazuje, jak ważne są badania nad przyczyną chorób. Gdyby bowiem nie intensywne badania nad SMA i białkiem SMN, którego brak powoduje to schorzenie, nie mielibyśmy leku – podkreśla prof. Kotulska.
Korzystaliśmy z informacji Dziennikarskiego Klubu Promocji Zdrowia, Fundacji SMA oraz serwisu Nauka i Zdrowie Polskiej Agencji Prasowej
/3 Rdzeniowy zanik mięśni – SMA – wywołuje mutacja w genie wytwarzającym białko niezbędne do działania ruchowych komórek nerwowych w rdzeniu kręgowym.
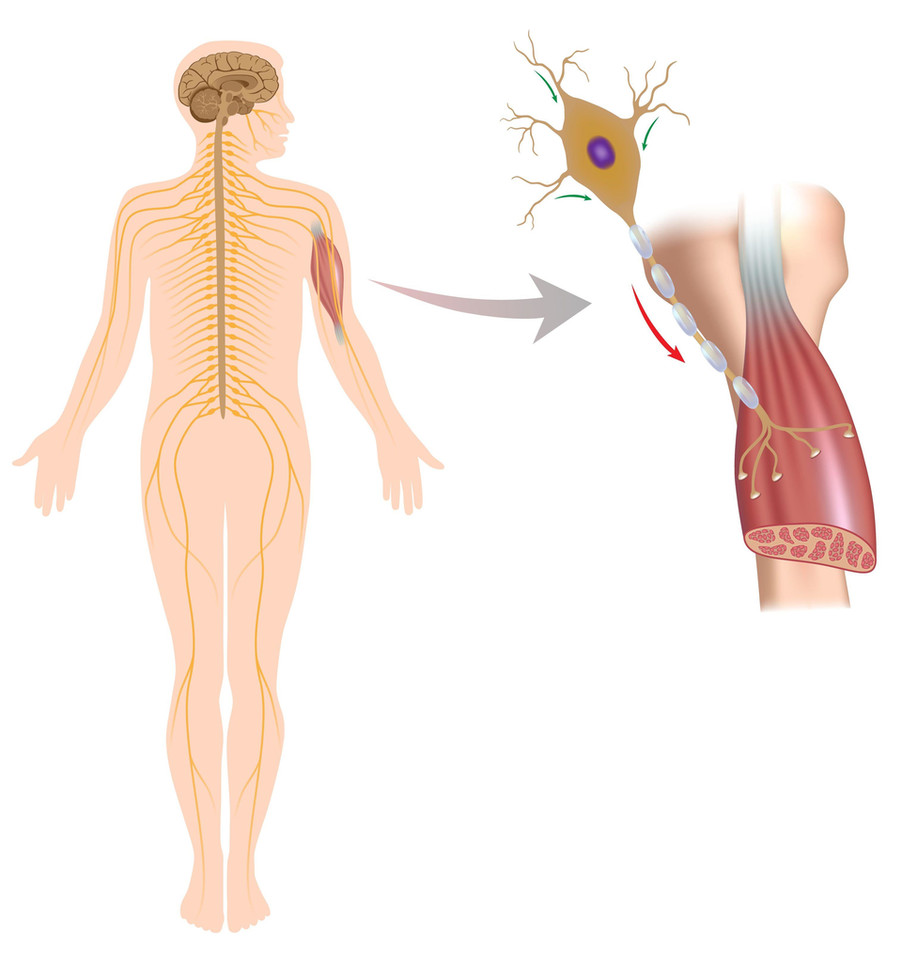
123RF
Kiedy tego białka zabraknie, neurony obumierają, a wraz z nimi słabną – i z czasem zanikają – mięśnie.
/3 O tym, czym jest rdzeniowy zanik mięśni, a także o nadziejach związanych z nowymi terapiami rozmawiali w Warszawie specjaliści oraz rodziny pacjentów z SMA.

Materiał prasowy
W spotkaniu wzięły udział, między innymi, prof. Katarzyna Kotulska–Jóźwiak – kierownik Kliniki Neurologii i Epileptologii w Centrum Zdrowia Dziecka oraz Kamila Górniak – współzałożycielka Fundacji SMA .
/3 Łańcuch DNA.

Autor: kk
źródło fakt.pl